Checkpointing (wavefunctions, moments, and reduced density)¶
Duo can checkpoint (save) and later reuse the following numerical objects: rovibronic eigenvalues and eigenvectors, vibrational basis functions on the radial grid, and (optionally) matrix elements of transition-moment operators (electric dipole, magnetic dipole, electric quadrupole, etc.).
This enables intensity calculations bypassing the eigenvalue problem completely. This functionality allows one to decouple the intensity production from the solution of the Schrödinger equation. This can be especially useful when the intensities for different values of 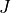 are processed simultaneously and potentially for refining dipole/quadrupole curves by fitting to experimental intensities.
are processed simultaneously and potentially for refining dipole/quadrupole curves by fitting to experimental intensities.
Other common uses of checkpointing are to generate reduced densities or to obtain expectation values.
The checkpointing workflow is controlled by the checkpoint input block.
Overview of the checkpoint block¶
Basic structure:
checkpoint
eigenfunc <save|read|none>
density <save|read>
dipole <save|read|calc>
filename <prefix>
end
The keyword filename defines the checkpoint prefix (for example YO), which is used to name the produced files; save, read, calc and none (not case sensitive) are allowed parameters defining the intended workflow.
The keyword eigenfunc (aliases eigenvectors and eigenvect) checkpoints eigenvalues, eigenvector coefficients, and the underlying vibrational basis functions on a grid of bond-length values. The latter two objects fully define the eigenvectors for any external use.
The keyword density controls the calculation and storage of reduced densities (see below).
What gets written: checkpoint files¶
When eigenfunc is set to save, the following files are stored:
<prefix>_values.chk(ascii) Rovibronic eigenvalues together with descriptors (quantum numbers, state labels, properties etc).<prefix>_vectors.chk(binary by default; optional ascii) Rovibronic eigenvector coefficients for all computed solutions (see Structure of <prefix>_vectors.chk (eigenvector checkpoint file)).<prefix>_vib.chk(ascii) Vibrational (contracted) basis functions tabulated on the radial grid.
When dipole is set to save, the following file is stored:
external.chk(binary)Stored vibrational matrix elements of transition moments (electric dipole, magnetic dipole and quadrupole) are saved. (Despite the generic name, this file belongs to the checkpoint dataset and should be kept together with the other
<prefix>_*files.)
Note
The exact set of operators stored in external.chk depends on what transition-moment curves are present in the input
and enabled in the calculation (e.g., electric dipole, magnetic dipole, quadrupole).
Writing checkpoints for later intensity work¶
A typical setup to compute energies/eigenvectors (and optionally moment matrix elements) and write them to disk is:
checkpoint
eigenfunc save
dipole save
filename YO
end
This writes YO_values.chk, YO_vectors.chk, YO_vib.chk, and external.chk.
Controlling the J-range that is saved¶
The range of values to be computed (and thus stored) can be defined either in the intensity block or via
the global jrot line at the top of the Duo input file.
Example using the intensity block:
intensity
absorption
states-only
J, 0.5, 6.5
freq-window -0.001, 20000.0
energy low -0.001, 3000.00, upper -0.00, 23000.0
end
Here, states-only tells Duo not to generate intensities during this run. The calculation still produces and stores
all rovibronic states for 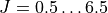 (as requested), which can then be reused later.
(as requested), which can then be reused later.
Alternatively, you can define the rotational range globally:
jrot 0.5 - 6.5
Tip
Using states-only together with checkpointing is a convenient way to generate and store states once, and perform
intensity calculations later (potentially split into multiple runs for different ).
Reading checkpoints to compute intensities (no recomputation)¶
Once the checkpoint data exist, Duo can compute intensities directly from the stored checkpoints.
Use:
checkpoint
eigenfunc read
dipole read
filename YO
end
and provide an intensity block for the transitions you want:
intensity
absorption
thresh_coeff 1e-60
thresh_dipole 1e-9
temperature 300.0
Qstat 337.0515
J, 5.5, 6.5
freq-window -0.001, 20000.0
energy low -0.001, 3000.00, upper -0.00, 23000.0
end
In this example, Duo reads the stored eigenvalues/eigenvectors/basis and transition-moment matrix elements and computes
intensities only for 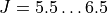 .
.
Main advantage: split and parallelise intensity production¶
Because intensities can be computed for subsets of (or different frequency/energy windows) from the same
checkpoint dataset, large spectra can be split into independent jobs, e.g.
compute and store states once for a broad
range;run multiple intensity jobs in parallel, each handling different
intervals or spectral windows;run “test” intensity calculations with different thresholds/windows without re-solving the Schrödinger equation.
Recomputing moment matrix elements with new dipole curves (dipole calc)¶
Another common use case is to keep eigenfunctions fixed while changing the transition-moment curves (e.g. refining an electric dipole moment function to experimental intensities).
In this mode Duo reads the stored wavefunctions/basis from checkpoint files, recomputes the transition-moment matrix elements:
checkpoint
eigenfunc read
dipole calc
filename YO
end
One would then run an intensity calculation using the updated matrix elements in the same run.
Controlling the format of *_vectors.chk (binary vs ascii)¶
By default Duo writes <prefix>_vectors.chk as a binary unformatted file (for speed and compactness). Duo can also
write an ascii version, which is larger but human-readable and convenient for debugging or post-processing.
The format is controlled by an optional selector after eigenvectors save (alias of eigenfunc save):
Binary (explicit):
checkpoint
eigenvectors save bin
filename O2_01
end
Binary (default; bin may be omitted):
checkpoint
eigenvectors save
filename O2_01
end
Ascii:
checkpoint
eigenvectors save ascii
filename O2_01
end
Note
The keywords are case-insensitive. The canonical spelling used in this manual is lowercase.
Structure of <prefix>_vectors.chk (eigenvector checkpoint file)¶
The file <prefix>_vectors.chk stores the eigenvector coefficients (expansion coefficients in the contracted basis)
for all computed values and symmetry blocks. The file also contains a copy of the contracted basis
quantum-number table so that eigenvectors can be interpreted externally.
Binary file layout (unformatted)¶
The binary *_vectors.chk is written as Fortran unformatted records. Conceptually, it consists of:
a header with a start marker and the saved
range,the contracted basis definition (one record per basis function),
a list of symmetry blocks, each containing all saved roots and their coefficient vectors,
an end marker.
The record sequence is:
Record 1: start marker (string)
Record 2:
nJ, jmin, jmax(integers/reals as used internally)Record 3:
Ntotal(integer; total size of the contracted basis)Records 4..(3+Ntotal): contracted-basis quantum numbers for each basis function
Record (after basis list): end marker for the basis table (string)
- For each saved rotational block and symmetry irrep:
Record:
J, irrep- For each saved root within this block:
Record:
total_roots, vec(running root index and coefficient vector)
Final record: end marker (string)
The implementation in diatomic.f90 follows the pattern:
open(unit=iunit, form='unformatted', action='write', status='replace', file=filename)
write(iunit) 'eigenvectors-chk/start'
write(iunit) nJ, jmin, jmax
write(iunit) Ntotal
do k = 1, Ntotal
write(iunit) icontr(k)%istate, icontr(k)%v, icontr(k)%ilambda, &
icontr(k)%spin, icontr(k)%sigma, icontr(k)%omega, icontr(k)%iomega, &
icontr(k)%ivib, icontr(k)%ilevel
enddo
write(iunit) 'contr-basis/end'
total_roots = 0
do irot = 1, nJ
do irrep = 1, sym%NrepresCs
write(iunit) J_list(irot), irrep
do i = 1, Nroots
total_roots = total_roots + 1
write(iunit) total_roots, vec
enddo
enddo
enddo
write(iunit) 'eigenvectors-chk/end'
Contracted-basis quantum numbers¶
Each basis-function record stores the quantum numbers and identifiers needed to interpret the coefficient vectors:
istateelectronic state indexvvibrational quantum labelilambda (integer; stored as used internally)
(integer; stored as used internally)spintotal electronic spin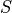 (real; e.g. 1.0 for triplet)
(real; e.g. 1.0 for triplet)sigma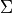 (real; projection of )
(real; projection of )omega (real; projection of on the molecular axis)
(real; projection of on the molecular axis)iomegainteger representation/index of used internallyivibvibrational basis index used internallyilevellevel/basis identifier used internally
Note
The exact meaning of ivib and ilevel is internal bookkeeping, but they are retained so that the saved vectors can
be mapped back onto the same contracted basis during eigenfunc read and for external post-processing.
Eigenvector blocks¶
For each  block, Duo writes
block, Duo writes Nroots eigenvectors. Each vector is stored as a 1D array
vec(1:Ntotal) containing the expansion coefficients in the contracted basis listed above.
The integer total_roots is a running counter over all saved roots, increasing across all and irreps.
Ascii file layout¶
In ascii mode, *_vectors.chk is a plain-text file with:
a header line giving the
range,a basis table (one line per basis function),
repeated blocks for each
and symmetry, containing each root and its vector components.
A typical structure is:
J range: 0.0 1.0
basis set quantum numbers/start
State v ilambda spin Sigma Omega
1 0 0 1.0 0.0 0.0
1 1 0 1.0 0.0 0.0
1 2 0 1.0 0.0 0.0
...
basis set quantum numbers/end
J = 0.0 Symmetry = 1
Root = 1 Size = 40
-0.999999998587E+00
-0.529926483660E-04
0.410237753030E-05
...
Root = 2 Size = 40
0.529923359425E-04
-0.999999995749E+00
...
Here, Size is the length of each vector (i.e. the contracted basis size for this run, typically Ntotal). Each
root block then lists exactly Size floating-point numbers (one coefficient per line).
Example: structure of *_values.chk (eigenvalue checkpoint file)¶
Below is an excerpt from KH_values.chk (KH project) illustrating the structure of an eigenvalues-checkpoint file:
Molecule = K H
masses = 38.963706486430 1.007825032230
Nroots = 20
Nbasis = 160
Nestates = 4
Npoints = 501
range = 0.9000000 16.0000000
nJs = 11
Jrange = 0.00000 10.00000
X1Sigma+, A1Sigma+, B1Pi, C1Sigma+, <- States
140 <- Nlevels
140 <- Ndimen
1 492.2960321790 0.0 1 1 X1Sigma+ 0 0 0.0 0.0 0.0 1 1 T 2.20122 0.11673E-29
2 1448.1762331140 0.0 1 1 X1Sigma+ 1 0 0.0 0.0 0.0 1 1 T 2.26446 0.97793E-30
3 2374.1127556956 0.0 1 1 X1Sigma+ 2 0 0.0 0.0 0.0 1 1 T 2.32626 0.32747E-29
4 3271.0297066643 0.0 1 1 X1Sigma+ 3 0 0.0 0.0 0.0 1 1 T ...
...
End of eigenvalues
The first ~10 lines form a fingerprint of the calculation (molecule, masses, basis size, grid, state list, etc.). When reading checkpoints, Duo checks it against the current input to ensure that checkpoint files are not accidentally reused across different models, parameter sets, grids, or molecules.
Reduced density calculations¶
In addition to wavefunction/moment checkpointing, Duo can compute and store the vibrational reduced density on the radial grid. This is enabled via:
checkpoint
density save
filename <prefix>
end
The reduced density 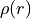 is computed as:
is computed as:
![\rho(r) =
\sum_{v,v'} \sum_{\mathrm{State},\Lambda,\Sigma}
\left[C_{v,\mathrm{State},\Lambda,\Sigma}^{(i)}\right]^*
C_{v',\mathrm{State},\Lambda,\Sigma}^{(i)}
\,\phi_v(r)^* \phi_{v'}(r)\,\Delta r .](_images/math/d1d8bdeee03d3aa966747ab3b8e1bba37dacf051.png)
A typical density checkpoint record has the following form:
0.545190480438E-08 || 1.5 0 1
0.286121234769E-07 || 1.5 0 1
0.134835397210E-06 || 1.5 0 1
...
The first column is the density value at grid point 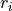 , followed by delimiter
, followed by delimiter ||, total angular momentum
[2], parity 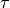 , and the state number (as in the Duo output).
, and the state number (as in the Duo output).
Notes on terminology and legacy keywords¶
Older inputs and parts of the manual may refer to eigenvectors save or use default prefixes such as eigen_*.
In the current syntax, the recommended form is:
eigenfunc save/eigenfunc readfor wavefunction checkpointingdipole save/dipole read/dipole calcfor moment matrix elementsfilename <prefix>to control output names
Footnote
Keywords¶
- ascii¶
Format selector for
eigenvectors save. Requests writing<prefix>_vectors.chkin plain text (human-readable) format.Example:
checkpoint eigenvectors save ascii filename o2_01 end
- bin¶
Format selector for
eigenvectors save. Requests writing<prefix>_vectors.chkas a binary unformatted file. This is the default: if neither selector is provided, Duo writes the binary format.Examples:
checkpoint eigenvectors save bin filename o2_01 end
checkpoint eigenvectors save filename o2_01 end
- checkpoint¶
Starts a checkpointing input block controlling saving/reading of wavefunctions, reduced densities, and (optionally) transition-moment matrix elements.
Basic form:
checkpoint eigenfunc <save|read|none> density <save|read> dipole <save|read|calc> filename <prefix> end
- density¶
Controls checkpointing of reduced densities on the radial grid. Allowed values are
saveandread.Examples:
density save
density read
- dipole¶
Controls checkpointing and reuse of transition-moment matrix elements (electric dipole, magnetic dipole, electric quadrupole, etc.). Allowed values are
save,read, andcalc.savewrites moment matrix elements toexternal.chk.readreuses matrix elements fromexternal.chk.calcrecomputes matrix elements in the current run (while typically reusing wavefunctions viaeigenfunc read).
Examples:
dipole save
dipole read
dipole calc
- eigenfunc¶
Controls checkpointing of rovibronic eigenvalues/eigenvectors and the vibrational basis needed to reconstruct eigenfunctions. Allowed values are
save,read, andnone.Notes: *
eigenvectorsandeigenvectare supported aliases. * Whensaveis requested, Duo writes<prefix>_values.chk,<prefix>_vectors.chkand<prefix>_vib.chk.Examples:
eigenfunc save
eigenfunc read
eigenfunc none
- eigenvectors¶
Alias of eigenfunc. In particular,
eigenvectors savesupports optional format selectors ascii and bin for the*_vectors.chkfile (see below).- filename¶
Sets the checkpoint dataset prefix
<prefix>used to name checkpoint files such as<prefix>_values.chk,<prefix>_vectors.chk, and<prefix>_vib.chk.Example:
filename o2_01
- none¶
Used with eigenfunc to disable eigenfunction checkpointing in the current run.
- read¶
Used with checkpoint-controlled quantities (e.g. eigenfunc, density, dipole) to request reading the corresponding checkpoint files and reusing stored data.
- save¶
Used with checkpoint-controlled quantities (e.g. eigenfunc, density, dipole) to request writing the corresponding checkpoint files.