Duo¶
Duo is a computer program for the spectroscopy of diatomic molecules. See Yurchenko et. al, Comput. Phys. Commun., 202, 262 (2016)
Its main functionalities belong to one of these three tasks:
Given a set of potential energy curves (PECs) Duo can solve the corresponding one-dimensional Schroedinger equation, which for
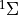 states is
states is![-\frac{\hbar^2}{2 \mu} \frac{\mathrm{d}^2}{\mathrm{d}r^2}\psi_{\upsilon J}(r) + \left[V_{\rm state}(r) + \frac{J(J+1)}{2 \mu r^2} \right] \psi_{\upsilon J}(r) = E_{\upsilon J} \psi_{\upsilon J}(r)](_images/math/797036703ddf55a7a0a09c5a2b3b4734823a337c.png)
and find the bound-state energies and wave functions. PECs may be coupled to one another by a variety of coupling terms, in which case the relevant coupling curves should be also provided. Supported couplings include spin-orbit, spin-electronic, spin-rotational, L-uncoupling and S-uncoupling.
Given a set of PECs, coupling curves and dipole moment curves Duo can compute line intensitie for rotational, vibrational and electronic transitions.
Given a set of reference energy levels or line positions (e.g., obtained from experiment) Duo can find the PECs and coupling curves which best reproduce the given data (empirical refinement of PECs or fitting).
Duo inputs can be broken down into three sections:
Calculation setup.
Specification of the Hamiltonian (PECs and couplings).
Calculation of spectra or fitting of PECs and couplings (optional).