Omega representation¶
Duo traditionally works in a Hund’s case (a) electronic basis, 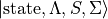 , i.e. the
, i.e. the Lambda–S (or vib) contraction. Duo can now also work in an 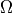 -based contracted representation, where the electronic + spin–orbit problem is diagonalised at each bond length and the resulting -labelled channels are used for vibrational contraction and for the final rovibronic basis.
-based contracted representation, where the electronic + spin–orbit problem is diagonalised at each bond length and the resulting -labelled channels are used for vibrational contraction and for the final rovibronic basis.
The 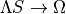 transformation (often called the state-interacting method) has been widely used to simplify the treatment of spin–orbit coupling in rovibronic calculations; see, e.g., :cite:p:21BeJoLia.SO`, [3], [4]. The key idea is to diagonalise the electronic Hamiltonian together with the Breit–Pauli spin–orbit Hamiltonian to obtain effective potentials for each spin–orbit component. While this can make the problem look “single-state-like”, strict equivalence with the original
transformation (often called the state-interacting method) has been widely used to simplify the treatment of spin–orbit coupling in rovibronic calculations; see, e.g., :cite:p:21BeJoLia.SO`, [3], [4]. The key idea is to diagonalise the electronic Hamiltonian together with the Breit–Pauli spin–orbit Hamiltonian to obtain effective potentials for each spin–orbit component. While this can make the problem look “single-state-like”, strict equivalence with the original 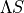 formulation requires
transforming the full nuclear-motion Hamiltonian, which introduces spin–orbit-induced non-adiabatic couplings (NACs); see, e.g., [5], [6], [7].
formulation requires
transforming the full nuclear-motion Hamiltonian, which introduces spin–orbit-induced non-adiabatic couplings (NACs); see, e.g., [5], [6], [7].
Transforming to the representation¶
A general workflow for building the representation is:
Solve the electronic Schrödinger equation to obtain electronic wavefunctions, and construct potential energy curves and spin–orbit coupling curves for the electronic states of interest.
Build, at each bond length
 , the electronic + spin–orbit Hamiltonian matrix
, the electronic + spin–orbit Hamiltonian matrix 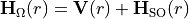 in the chosen electronic basis.
in the chosen electronic basis.Diagonalise
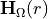 at each to obtain spin–orbit-decoupled channels and effective potentials labelled by .
at each to obtain spin–orbit-decoupled channels and effective potentials labelled by .To achieve exact equivalence with the original
representation, apply the same r-dependent unitary transformation to the remaining parts of the rovibronic Hamiltonian. In particular, transforming radial derivative operators generates NAC terms that must be included for accurate energies, wavefunctions, and intensities.
The diatom in the and representations¶
In a Hund’s case (a) basis, the coupled rovibronic Schrödinger equation can be written as (see also the standard Duo theory in the manual):
(1)¶![\left[
\frac{\hbar^2}{2\mu}\left(-\frac{d^2}{dr^2} + \frac{1}{r^2}\hat{\mathbf{R}}^2\right)
+ \mathbf{V}(r) + \mathbf{H}_{\rm SO}(r)
\right]\vec{\chi}(r) = E_i\,\vec{\chi}(r),](_images/math/167836f68f734669e2d137d1ef656e45e697ebf8.png)
where 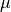 is the reduced mass, is the internuclear separation,
is the reduced mass, is the internuclear separation, 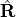 is the nuclear rotational angular momentum operator,
is the nuclear rotational angular momentum operator, 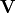 contains diagonal Born–Oppenheimer PECs [8], and
contains diagonal Born–Oppenheimer PECs [8], and 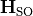 contains spin–orbit matrix elements (e.g. from ab initio electronic-structure calculations).
contains spin–orbit matrix elements (e.g. from ab initio electronic-structure calculations).
State-interacting diagonalisation¶
The state-interacting transformation is defined by diagonalising 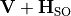 at each :
at each :
(2)¶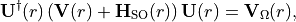
where 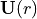 is an -dependent unitary matrix and
is an -dependent unitary matrix and 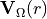 is diagonal. This defines the transformation of the electronic basis
is diagonal. This defines the transformation of the electronic basis
(3)¶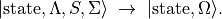
It is tempting to assume that yields fully decoupled single-channel rovibronic problems. However, to remain formally consistent, the kinetic-energy operators in Eq. (1) must also be transformed. The transformation of radial derivatives introduces NAC terms.
Spin–orbit-induced NACs from the vibrational kinetic energy¶
Upon transforming the radial kinetic-energy operator, one obtains additional terms of the standard adiabatic/NAC form (see diabatisation theory, e.g. [9], [10], [6], [7]):
(4)¶![-\frac{\hbar^2}{2\mu}\mathbf{U}^\dagger \frac{d^2}{dr^2}\mathbf{U}
=
-\frac{\hbar^2}{2\mu}\left[
\frac{d^2}{dr^2}
+ \mathbf{W}^2
-\left(\frac{d}{dr}\mathbf{W} - \mathbf{W}\frac{d}{dr}\right)
\right],](_images/math/37dbb7f6ac98e8e1b50947021eb7e498b0f27c87.png)
where
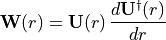
is a skew-Hermitian matrix of derivative couplings. The diagonal elements of 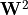 act as additional diagonal corrections (analogous in spirit to DBOC-like terms), while off-diagonal terms mediate non-adiabatic transitions between channels of the same .
act as additional diagonal corrections (analogous in spirit to DBOC-like terms), while off-diagonal terms mediate non-adiabatic transitions between channels of the same .
How to use the Omega representation in Duo¶
Switching from the standard Lambda–S contraction to the contraction requires only a change in the
contraction block:
contraction
omega
vmax 20 40 40
end
Here the usual vib (Lambda–S) option is replaced by omega.
High-level algorithm used by Duo (omega mode)¶
When omega is selected, Duo performs (conceptually) the following steps:
At each radial grid point
, build an extended electronic matrix and diagonalise it to obtain and . Duo then uses to transform relevant curve-based operators to the representation and to generate the induced NAC terms arising from the -dependence of .Note
The “extended matrix” used for diagonalisation includes spin–orbit together with other J-independent electronic terms available in the model. Any terms not included at this stage are still handled in the full rovibronic Hamiltonian, but the precise partitioning of terms between “diagonalisation” and “final Hamiltonian” affects the structure of the induced NACs and should be treated consistently.
Solve the pure vibrational problems for each
channel using the chosen DVR method (e.g. sinc DVR), obtaining vibrational functions 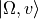 .
.Use these vibrational functions to compute vibrational matrix elements of all required operators and curves (PECs, DMCs, couplings) in the
representation.Construct the final rovibronic basis and Hamiltonian. Schematically,
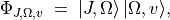
which are then symmetrised (Wang functions) and used to build and diagonalise the rovibronic Hamiltonian.
If requested, compute rovibronic intensities and/or line lists using the same workflows as in the
Lambda–Smode.
Omega-specific output¶
The output is similar to the standard Lambda–S mode, but includes additional diagnostics for curves and operators transformed to the representation. For example, Duo can print PECs labelled by :
PECS in the Omega representation
# # Omega Lambda Sigma State
1 1 -1.0 0 -1.0 a3Sigma-
2 1 0.0 0 0.0 X1Sig+
...
and transformed multi-component operators such as the electronic spin operator (schematically shown here):
S = (S+,S-) in the Omega representation
# i Omega State Lambda Sigma <-> j Omega State Lambda Sigma
1 1 -1.0 2 0 -1.0 1 0.0 1 0 0.0
...
Similarly, dipole moment curves can be printed in the representation:
Dipole in the Omega representation
# Omega State Lambda Sigma Omega State Lambda Sigma
1 0.0 1 0 0.0 0.0 1 0 0.0
...
Warning and words of caution¶
Warning
The representation in Duo is currently a work in progress. It has been tested on a limited set of systems and couplings (see [11]) and may produce incorrect results for models involving additional couplings or corrections not yet fully validated in omega mode. If you use omega mode beyond the tested scope, it is strongly recommended to validate against the standard Lambda–S calculation.
Even when technically available, the fully decoupled (single-state) approximation should not be used blindly. Transforming to the representation unavoidably introduces induced NAC terms which may be essential for accurate energies and transition properties, especially for spin-forbidden bands. The broader lesson is that unitary “simplifications” must be accompanied by an accounting of the physics moved elsewhere in the Hamiltonian (see discussion and examples in [11]).
Keywords¶
- omega¶
Option in the
contractionblock that requests contraction and rovibronic calculations in the representation. This triggers an -dependent diagonalisation to define channels and includes the induced non-adiabatic couplings required for formal consistency.Example:
contraction omega vmax 20 40 40 end
- vib¶
Standard option in the
contractionblock for the Hund’s case (a)Lambda–Srepresentation (the default behaviour in Duo). In this manual it may also be referred to as theLambda–Scontraction.Example:
contraction vib vmax 20 40 40 end
References¶
Hans-Joachim Werner, Peter J. Knowles, Frederick R. Manby, Joshua A. Black, Klaus Doll, Andreas Heßelmann, Daniel Kats, Andreas Köhn, Tatiana Korona, David A. Kreplin, Qianli Ma, Thomas F. Miller, Alexander Mitrushchenkov, Kirk A. Peterson, Iakov Polyak, Guntram Rauhut, and Marat Sibaev. The Molpro quantum chemistry package. J. Chem. Phys., 152:144107, 2020. doi:10.1063/5.0005081.
Christel M. Marian. Spin-orbit coupling in molecules. In K B Lipkowitz and D B Boyd, editors, Rev. Comput. Chem., volume 17, pages 99–204. John Wiley & Sons, Inc., 2001.
Pavel Pokhilko, Evgeny Epifanovsky, and Anna I. Krylov. General framework for calculating spin–orbit couplings using spinless one-particle density matrices: Theory and application to the equation-of-motion coupled-cluster wave functions. J. Chem. Phys., 151:034106, 2019. doi:10.1063/1.5108762.
Le Yu and Wensheng Bian. Extensive Theoretical Study on Electronically Excited States and Predissociation Mechanisms of Sulfur Monoxide Including Spin-Orbit Coupling. J. Comput. Chem., 32:1577–1588, 2011. doi:10.1002/jcc.21737.
M. Tamanis, I. Klincare, A. Kruzins, O. Nikolayeva, R. Ferber, E. A. Pazyuk, and A. V. Stolyarov. Direct excitation of the “dark” $b$ $^3\Pi $ state predicted by deperturbation analysis of the $A$ $^1\Sigma ^+$–$b$ $^3\Pi $ complex in KCs. Phys. Rev. A, 82:032506, 2010. doi:10.1103/PhysRevA.82.032506.
Ryan P. Brady, Charlie Drury, Sergei N. Yurchenko, and Jonathan Tennyson. Numerical Equivalence of Diabatic and Adiabatic Representations in Diatomic Molecules. J. Chem. Theory Comput., 20:2127–2139, 2024. doi:10.1021/acs.jctc.3c01150.
R. P. Brady. A strict and internally consistent diabatic representation for coupled N-state diatomics: a hybrid asymptotic-property-based diabatization method. J. Chem. Phys., 162:174105, 2025. doi:10.1063/5.0260594.
M. Born and R. Oppenheimer. Zur Quantentheorie der Molekeln. Annalen der Physik, 389(20):457–484, 1927. doi:10.1002/andp.19273892002.
Michael Baer. Topological effects in molecular systems: an attempt towards a complete theory. Chem. Phys., 259(2-3):123–147, sep 2000. doi:10.1016/S0301-0104(00)00193-2.
Michael Baer and Alexander Alijah. Quantized non-adiabatic coupling terms to ensure diabatic potentials. Chem. Phys. Lett., 319(5-6):489–493, mar 2000. doi:10.1016/S0009-2614(00)00195-0.
Ryan P. Brady and Sergei N. Yurchenko. Spin-orbit induced non-adiabatic dynamics: an exact ω-representation. 2026. URL: https://arxiv.org/abs/2603.06306, arXiv:2603.06306.
Sergei N. Yurchenko, Lorenzo Lodi, Jonathan Tennyson, and Andrey V. Stolyarov. Duo: a general program for calculating spectra of diatomic molecules. Comput. Phys. Commun., 202:262 – 275, 2016. URL: http://www.sciencedirect.com/science/article/pii/S0010465516000023, doi:http://dx.doi.org/10.1016/j.cpc.2015.12.021.
Laxmi Prajapat, Pawel Jagoda, Lorenzo Lodi, Maire N. Gorman, Sergei N. Yurchenko, and Jonathan Tennyson. ExoMol molecular line lists - XXIII. Spectra of PO and PS. MNRAS, 472:3648–3658, 2017. URL: http://dx.doi.org/10.1093/mnras/stx2229, doi:10.1093/mnras/stx2229.
Sergei N Yurchenko, Emma Nogué, Ala'a A A Azzam, and Jonathan Tennyson. ExoMol line lists - XLVII. Rovibronic spectrum of aluminium monochloride (AlCl). MNRAS, 520(4):5183–5191, 2022. doi:10.1093/mnras/stac3757.
Qianwei Qu, Sergei N. Yurchenko, and Jonathan Tennyson. A variational model for the hyperfine resolved spectrum of VO in its ground electronic state. J. Chem. Phys., 157:124305, 2022. doi:10.1063/5.0105965.
W. Somogyi, S. N. Yurchenko, and A. Yachmenev. Calculation of electric quadrupole linestrengths for diatomic molecules: Application to the H$_2$, CO, HF, and O$_2$ molecules. J. Chem. Phys., 155:214303, 2021. doi:10.1063/5.0063256.
Wilfrid Somogyi, Sergey N. Yurchenko, and Gap-Sue Kim. An \textit ab initio spectroscopic model of the molecular oxygen atmospheric and infrared bands. Phys. Chem. Chem. Phys., 26:27419–27430, 2024. doi:10.1039/D4CP02619E.
Mikhail Semenov, Sergei. N. Yurchenko, and Jonathan Tennyson. Predicted lande g-factors for open shell diatomic molecules. J. Mol. Spectrosc., 330:57–62, 2016. doi:10.1016/j.jms.2016.11.004.