Duo fitting¶
Duo can refine (fit) potential energy curves (PECs) and coupling curves by least-squares fitting to experimental term values (energies) or wavenumbers (line positions).
Duo uses a Gauss–Newton least-squares procedure with Marquardt damping. Optionally, this can be combined with a simple line search (linear_search).
The linear least-squares problem is solved using either the LAPACK routine DGELSS or Duo’s internal solver LINUR, controlled by fit_type
(aliases: fit_type, fit-type, FIT_TYPE).
Fitting is the most delicate part of a Duo workflow. While forward calculations (energies and spectra for fixed PECs/couplings/dipoles) are usually straightforward-provided the phase conventions for coupling terms are consistent-fitting often requires careful data selection and some trial and error. It is also an area where further development is expected in future versions.
Example¶
A minimal example of a fitting section is:
FITTING
JLIST 2.5, 0.5, 1.5 - 11.5, 22.5 - 112.5
itmax 30
fit_factor 1e6
output alo_01
fit_type dgelss
fit_scale 0.001
linear_search 5
lock 5.0
robust 0.001
energies (J parity NN energy) (e-state v ilambda isigma omega weight)
0.5 + 1 0.0000 1 0 0 0.5 0.5 100.000
0.5 + 2 965.4519 1 1 0 0.5 0.5 7.071
0.5 + 3 1916.8596 1 2 0 0.5 0.5 5.774
0.5 + 4 2854.2366 1 3 0 0.5 0.5 5.000
0.5 + 5 3777.5016 1 4 0 0.5 0.5 4.472
0.5 + 6 4686.7136 1 5 0 0.5 0.5 4.082
0.5 + 7 5346.1146 2 0 1 -0.5 0.5 100.000
end
State labels in the FITTING section¶
If electronic states are defined using labels (instead of numeric indices), e.g.
potential X
...
end
potential A
...
end
then the same labels should be used in the energies / frequencies tables:
FITTING
JLIST 2.5, 0.5, 1.5 - 11.5, 22.5 - 112.5
itmax 30
fit_factor 1e6
lock 5.0
energies (J parity NN energy) (e-state v ilambda isigma omega weight)
0.5 + 1 0.0000 X 0 0 0.5 0.5 100.000
0.5 + 2 965.4519 X 1 0 0.5 0.5 7.071
0.5 + 3 1916.8596 X 2 0 0.5 0.5 5.774
0.5 + 6 4686.7136 X 5 0 0.5 0.5 4.082
0.5 + 7 5346.1146 A 0 1 -0.5 0.5 100.000
end
Note
If you use the states option to restrict the electronic states, it is recommended to keep the lowest (ground) PEC included, even if it is not used
directly in the fit. Otherwise Duo may not be able to determine a consistent zero-point energy (ZPE) shift, leading to very large obs.–calc residuals.
Fitting ranges (parameter bounds)¶
Bounds on fitted parameters can be specified using the keyword range on the same line as the parameter:
poten Ap
name "Ap2Delta"
lambda 2
mult 2
type EHH
values
V0 1.47076002476226e+04 fit
RE 1.81413234392416e+00 fit range 1.7, 1.9
DE 5.92200000000000E+04 link 1,2,3
ALPHA 1.51288898501269E+00 fit
C 6.54666674267795E-03 fit
B1 -2.63874034064552E+00 fit
B2 -4.80709545680641E+00 fit
B3 0.000000000000000000
end
The keywords fit and range are complementary; their order on the line is not important as long as they appear after the parameter value.
Conceptually, the bound enforces
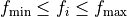 at each iteration:
.. math:
at each iteration:
.. math:
\begin{split}
f_i &\leftarrow \max(f_i, f_{\rm min}), \\
f_i &\leftarrow \min(f_i, f_{\rm max}).
\end{split}
Keywords¶
- energies¶
Starts a table of experimental term values to be fitted.
Example:
energies 0.5 + 1 0.0000 1 0 0 0.5 0.5 1.00 0.5 + 2 965.4519 1 1 0 0.5 0.5 0.90 0.5 + 3 1916.8596 1 2 0 0.5 0.5 0.80 end
Column meanings:
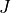 - total angular momentum quantum number.
- total angular momentum quantum number.parity - either total parity
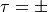 or
or 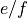 parity.
parity.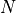 - running number counting levels in ascending order of energy
within a
- running number counting levels in ascending order of energy
within a 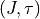 symmetry block.
symmetry block.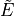 - term value (cm-1).
- term value (cm-1).state - electronic state index/label as defined in
potential. - vibrational quantum number.
- vibrational quantum number.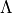 - projection of electronic orbital angular momentum
(integer; matched by absolute value).
- projection of electronic orbital angular momentum
(integer; matched by absolute value).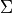 - projection of electronic spin (integer or half-integer;
matched by absolute value).
- projection of electronic spin (integer or half-integer;
matched by absolute value).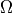 - projection of total electronic angular momentum (integer
or half-integer; matched by absolute value).
- projection of total electronic angular momentum (integer
or half-integer; matched by absolute value).weight - typically
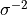 , where
, where 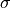 is the
experimental uncertainty.
is the
experimental uncertainty.
- fit_factor¶
Controls the relative importance of experimental data versus reference (
abinitio) constraints by scaling the experimental fitting weights.Large values (e.g.
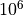 ) weaken the constraint to the reference
curve, so the fit primarily targets experimental obs.–calc residuals.
) weaken the constraint to the reference
curve, so the fit primarily targets experimental obs.–calc residuals.Small values (e.g.
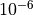 ) enforce close agreement with the
reference curve.
) enforce close agreement with the
reference curve.Extremely small values (below
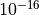 ) effectively ignore
experimental data (fit to the reference only).
) effectively ignore
experimental data (fit to the reference only).
Example:
fit_factor 1e2
- fit_scale¶
Fixed scaling factor
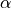 applied to the parameter update
applied to the parameter update
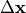 (a constant alternative to line search).
(a constant alternative to line search).This is ignored when a line search is active.
Example:
fit_scale 0.5
- fit_type¶
Chooses the linear solver used in the Gauss–Newton step.
Supported values:
DGELSS(LAPACK; often more stable for strongly correlated problems)LINUR(internal; can be faster in some cases)
Examples:
fit_type dgelss fit_type linur
- FITTING¶
Marks the beginning of the fitting input section.
The whole section can be deactivated by writing
FITTING offorFITTING none. This is useful to disable fitting without removing the block from the input file.- frequencies¶
(Aliases:
frequency,wavenumbers)Starts a table of experimental line positions (cm-1) to be fitted.
Example:
frequencies 0.0 + 2 0.0 + 1 720.0000 2 0 1 -1.0 0.5 1 0 0 0.0 0.0 1.00 2.0 + 17 3.0 - 2 5638.1376 4 0 0 1.0 1.0 2 0 -1 -1.0 -2.0 1.00 4.0 + 17 5.0 - 2 5627.5270 4 0 0 1.0 1.0 2 0 -1 -1.0 -2.0 1.00 4.0 + 18 7.0 - 2 5616.7976 4 0 0 0.0 0.0 2 0 -1 -1.0 -2.0 1.00 end
The quantities are (upper/lower indicated by prime/double prime):
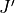 ,
, 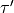 ,
, 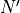 ,
, 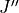 ,
, 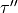 ,
,
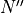 ; frequency (cm-1); state
; frequency (cm-1); state ,
, 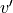 ,
,
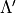 ,
, 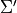 ,
, 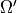 ; state
; state ,
,
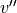 ,
, 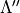 ,
, 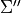 ,
, 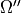 ; weight.
; weight.- itmax¶
(Alias:
itermax)Maximum number of fitting iterations.
Setting
itmax 0performs a straight-through calculation (no parameter updates), but prints obs.–calc residuals.Example:
itmax 15
- jlist¶
(Aliases:
jrot,J)Specifies which values of the total angular momentum quantum number
are used in the fit. Individual values can be separated by
spaces or commas; ranges are specified by a hyphen (hyphens should be
surrounded by spaces). The first value in jlistis used to determine the zero-point energy (ZPE) reference for fitting output.Example:
JLIST 1.5, 5.5, 15.5 - 25.5, 112.5
- linear_search¶
Enables a damped Gauss–Newton line search for a scaling factor
applied to the parameter update: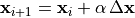 , with
, with
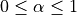 .
.Duo tests decreasing values starting from
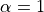 in steps of
in steps of
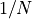 (Armijo condition), where
(Armijo condition), where Nis the integer argument.Example:
linear_search 5
- lock¶
(Alias:
thresh_assign)Assignment/locking threshold (cm-1).
When enabled, Duo uses quantum numbers (state,
,
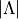 ,
,  ,
,  ) to identify and lock
levels/transitions within the threshold instead of relying only on the
running number within a block.
) to identify and lock
levels/transitions within the threshold instead of relying only on the
running number within a block.Missing or
lock 0: feature off (match by running number).lock > 0: match/lock by quantum numbers within the threshold.lock < 0: attempt matching using closest energies only (no label locking).
Example:
lock 20.0
- off¶
(Synonym:
none)Can be used to disable input blocks such as
FITTINGwhen placed next to the corresponding keyword, e.g.FITTING off.- output¶
Output prefix used for auxiliary per-iteration files:
<output>.en(term values / obs.–calc diagnostics)<output>.freq(line positions / obs.–calc diagnostics, if usingfrequencies)<output>.pot(residuals to reference curve, if present)
Example:
output NaH_fit
- range¶
Specifies bounds for a fitted parameter. The keyword
rangemust appear on the same line as the parameter value (typically afterfit), e.g.RE 1.81413234392416e+00 fit range 1.7, 1.9
The bounds enforce
during the
fit.- robust¶
Enables Watson’s robust fitting procedure.
robust 0: offrobust > 0: on, with the value setting the target weighted RMS error
Example:
robust 0.01
- target_rms¶
Convergence threshold (cm-1) for the unweighted RMS error.
Example:
target_rms 0.1
- thresh_obs-calc¶
Excludes data from the fit if the absolute obs.–calc residual exceeds the given threshold.
This can be useful when assignments swap repeatedly and
lockis insufficient. Default is 0 (off).
Structure of the fitting output¶
During fitting Duo prints per-iteration residuals. The first line below (numbers 1–20) is a legend only:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
1 1 0.5 + 0.0000 0.0000 0.0000 0.60E-02 1 0 1 -0.5 0.5 0.5 1 0 1 -0.5 0.5 0.5
2 2 0.5 + 1970.2743 1970.3983 -0.1240 0.59E-02 1 1 1 -0.5 0.5 0.5 1 1 1 -0.5 0.5 0.5
...
Column meanings:
line counter
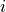 over all lines;
over all lines;counter
within each block;- ;
parity
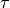 ;
;
5 - 6. observed and calculated value;
obs.–calc residual;
weight;
9 – 14. lower-state quantum numbers (state, , , , , 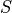 );
);
15 – 20. upper-state quantum numbers (same ordering as 9–14).
Auxiliary files: .en, .freq, .pot¶
The file <output>.en contains computed term values with assignments and (when
available) the experimental values, for each iteration. An asterisk * at the
end of a line indicates either a disagreement between theoretical and experimental
assignments or an obs.–calc residual larger than the lock threshold.
The file <output>.freq (when using frequencies) contains diagnostics for
the transitions listed in the input. For each listed transition Duo may also
report nearby candidate transitions whose upper/lower energies fall within
lock cm-1, which can help identify mis-assignments.
The file <output>.pot contains residuals between the fitted curve and its
reference (if an abinitio object is provided). This file is overwritten at
each iteration.
Fitting grid points¶
In addition to fitting analytic parameters, Duo can fit grid values for fields
given in a grid representation. Duo maps the input grid
(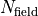 points) onto the internal Duo radial grid
(
points) onto the internal Duo radial grid
(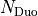 points) using cubic splines. The input grid values
points) using cubic splines. The input grid values
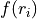 can therefore be treated as fit parameters.
can therefore be treated as fit parameters.
Example (fitting selected grid points of a spin–orbit curve):
spin-orbit 1 1
name "<X2Delta|SO|X2Delta>"
spin 0.5 0.5
lambda 2 2
sigma 0.5 0.5
type grid
factor 1.0 (sqrt2)
values
0.750000000000 -597.1915000 fit
1.200000000000 -590.2260000 fit
1.500000000000 -599.7850000 fit
2.000000000000 -603.9980000 fit
3.000000000000 -603.0000000 fit
end
Constrained fit to ab initio¶
If reference (abinitio) fields are provided, the fit can be constrained
towards these values. This is useful when experimental data are limited and the
fit would otherwise become under-determined.
The relative importance of experimental vs reference data is controlled by
fit_factor in the FITTING section (scaling experimental weights), and
optionally by fit_factor inside individual abinitio blocks.
Example:
FITTING
JLIST 2.5, 0.5, 1.5 - 11.5, 22.5 - 112.5
itmax 30
fit_factor 1e6
...
An abinitio block can also specify its own scaling:
abinitio poten X
name "X2Sigma"
lambda 0
symmetry +
mult 2
fit_factor 0.001
type grid
values
1.400000 40782.9118
1.450000 28672.6462
...
end
Note
The reference (abinitio) field does not have to be a grid field. Any
representation supported by Duo (including analytic forms) can be used as a
constraint.
Ab initio weights¶
Reference data must be weighted. This can be done by providing a third column of
weights in the values table:
abinitio poten X
name "X2Sigma"
lambda 0
symmetry +
mult 2
type grid
fit_factor 0.1
values
1.400000 40782.9118 0.5
1.450000 28672.6462 0.6
1.500000 19310.0950 0.7
1.550000 12244.8264 0.8
1.600000 7101.7478 0.9
1.650000 3562.3190 1.0
Alternatively, weights can be generated via weighting using the predefined function PS1997 (Partridge & Schwenke, 1997):
![w(r) = \frac{\tanh\!\left(-\beta\left[(V(r)-V_{\rm min})-V_{\rm top}\right] + 1.000020000200002\right)}{2.000020000200002}](_images/math/523bcac25aa1fef2c680936332dd84c593b83bc2.png)
where 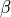 and
and 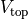 are parameters and
are parameters and 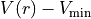 is the corresponding potential shifted to its minimum.
is the corresponding potential shifted to its minimum.
Example:
abinitio poten 1
name "X 2Pi"
lambda 1
mult 2
type grid
weighting ps1997 1e-3 20000.0
fit_factor 1e-8
values
0.6 610516.16994 0
0.7 294361.15182 0
end
The weighting feature can also be used for couplings. In that case,
is taken from the potential associated with the coupling.
For example:
abinitio spin-orbit-x A A
name "<A2Pi|LSZ|A2Pi>"
spin 0.5 0.5
lambda 1 1
sigma 0.5 0.5
factor -i 1.175
fit_factor 1e-2
weighting ps1997 0.0001 45000.0
type grid
<x|Lz|y> -i -i
values
1.58 163.05
1.59 163.82
...
end
For a non-diagonal coupling between states and 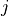 , Duo uses the potential of the first index () to define .
, Duo uses the potential of the first index () to define .
Example: refinement of the BeH PEC¶
This PEC can be refined by fitting to experimental energies using:
poten 1
name 'X2Sigma+'
lambda 0
symmetry +
mult 2
type EMO
values
V0 0.00
RE 1.342394
DE 17590.00 fit
RREF -1.00000000
PL 3.00000000
PR 3.00000000
NL 0.00000000
NR 0.00000000
b0 1.8450002 fit
end
FITTING
JLIST 0.5 - 0.5
itmax 12
fit_factor 1e5
output BeH_01
lock 1000
robust 0.0001
energies (state v ilambda isigma omega weight comment)
0.5 + 1 0.0000 1 0 0 0.5 0.5 1.00
0.5 + 2 1986.4160 1 1 0 0.5 0.5 1.00
...
end
A reference (ab initio) PEC can be used to constrain the fit:
abinitio poten 1
units cm-1 angstroms
name 'X2Sigma+'
lambda 0
symmetry +
mult 2
type grid
values
0.60 105169.63
0.65 77543.34
...
20.00 0.0
end