Specification of curves and couplings (Duo objects)¶
Once the main global parameters have been specified as described in the previous sections, it is necessary to introduce the PECs and the various coupling
curves defining the Hamiltonian. Dipole moment curves (DMCs), which are necessary for calculating spectral line intensities, are also discussed in this section, as well as some special objects which are used for fitting. Each object specification consists in a first part in which
keywords are given and a second one (starting from the values keyword) in which numerical values are given;
the order of the keywords is not important, except for values. Each object specification is terminated by the end keyword.
Objects of type poten (i.e., PECs, discussed in more detail below) begin with a line of the kind poten N where N is an integer index number counting over potentials and identifying them. It is recommended that PECs are numbered progressively as 1,2,3,…, although this only restriction is that the total number Nmax of PECs should be not less than the total number of states specified by the keywork nstates.
Most other objects (e.g., spin-orbit) are assumed to be matrix elements of some operator between electronic wave functions and after the keyword identifying their type require two integer numbers specifying the two indexes of the two electronic states involved (bra and ket). The indexes are the numbers specified after the texttt{poten} keyword.
Currently Duo supports the following types of objects: potential, spinorbit, L2, Lx, spinspin, spinspino, bobrot,
spinrot, diabatic, lambdaopq, lambdap2q, lambdaq, abinitio, brot, dipoletm, nac.
potential¶
Alias: poten. Objects of type poten represent potential energy curves (PECs) and are the most fundamental objects underlying each calculation. From the point of view of theory each PEC is the solution of the electronic Schoedinger equation with clamped nuclei, possibly complemented with the scalar-relativistic correction and with the Born-Oppenheimer Diagonal correction (also known as adiabatic correction). Approximate PECs can be obtained with well-known quantum chemistry methods such as Hartree-Fock, coupled cluster theory etc.
Objects of type poten or potential should always appear before all other objects as they are used to assign to each electronic states its quantum numbers. Here is an example for a PEC showing the general structure:
poten 1
name "a 3Piu"
symmetry u
type EMO
lambda 1
mult 3
values
V0 0.82956283449835E+03
RE 0.13544137530870E+01
DE 0.50061051451709E+05
RREF -0.10000000000000E+01
PL 0.40000000000000E+01
PR 0.40000000000000E+01
NL 0.20000000000000E+01
NR 0.20000000000000E+01
B0 0.20320375686486E+01
B1 -0.92543284427290E-02
B2 0.00000000000000E+00
end
Here poten 1 refers to the electronic state 1. This label 1 should be used consistently in all couplings as well as in the description of the experimental data.
From 2023, the state labels can be any string of characters, e.g.
poten Ap
name "Ap2Delta"
lambda 2
mult 2
type EMO
values
V0 1.47069212003828e+04 fit ( 1.47070955806154e+04)
RE 1.817000000000000000
DE 5.92200000000000E+04
RREF -1.00000000000000E+00
PL 4.00000000000000E+00
PR 4.00000000000000E+00
NL 1.00000000000000E+00
NR 4.00000000000000E+00
B0 1.700000000000000000
B1 0.000000000000000000
B2 0.000000000000000000
B3 0.000000000000000000
B4 0.000000000000000000
end
Integers 1,2,3 from before 2023 will continue working.
abinitio¶
Objects of type abinitio (aliases: reference, anchor) are reference, abinitio curves which may be specified during fitting. When they are used they constrain the fit so that the fitted function differs as little as possible from the ab initio (reference). The reference curve is typically obtained by ab initio methods. For any Duo object one can specify a corresponding reference curve as in the following example:
abinitio spin-orbit 1 2
name "<3.1,S=0,0 (B1pSigma)|LSX|+1 (d3Pig),S=1,1>"
spin 0.0 1.0
type grid
units bohr cm-1
values
2.3 -3.207178925 13.0
2.4 -3.668814404 24.0
2.5 -4.010985122 35.0
2.6 -4.271163495 46.0
2.7 -4.445721312 47.0
2.8 -4.468083270 48.0
end
Lx and L+¶
Aliases: Lplus, LxLy and L+. It represent matrix elements between electronic states of the molecule-fixed angular momentum operator 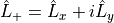 and
and 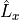 in the
in the  - and Cartesian-representations, respectively.
- and Cartesian-representations, respectively.
spin-orbit and spin-orbit-x¶
These objects are matrix elements of the Breit-Pauli spin-orbit Hamiltonian in the - and Cartesian-representations, respectively.
Example:
spin-orbit 1 3
name "<0,S=0 (X1Sigma+)|LSY|+1 (a3Pi),S=1> SO1"
spin 0.0 1.0
lambda 0 -1
sigma 0.0 -1.0
type grid
factor sqrt(2) (1 or i)
units bohr cm-1
values
2.80 17.500000
2.90 15.159900
3.00 12.347700
3.10 9.050780
3.20 5.391190
3.30 1.256660
3.40 -3.304040
3.50 -8.104950
3.60 -12.848400
3.70 -17.229100
3.80 -21.049000
3.90 -24.250400
4.00 -26.876900
4.10 -29.014700
4.20 -30.756100
4.30 -32.181900
4.50 -34.335500
5.00 -37.348300
end
Here 1 and 3 refer to the electronic states 1 and 3 as introduced using the corresponding potential:
potential 1
name . . .
. . .
end
and
potential 3
. . . . . .
end
From 2023, for the electronic states can be labelled using strings of characters, e.g.
spin-orbit-x A A
name "<A2Pi|LSZ|A2Pi>"
spin 0.5 0.5
lambda 1 1
sigma 0.5 0.5
units cm-1
factor -i (0, 1 or i)
type polynom_decay_24
<x|Lz|y> -i -i
values
RE 1.79280000000000E+00
BETA 8.00000000000000E-01
GAMMA 2.00000000000000E-02
P 6.00000000000000E+00
B0 2.06176847388046e+02
B1 -7.04066795005532e+01
B2 0.000000000000000000
B3 0.00000000000000E+00
BINF 220.0
end
where A is the reference label used for the electronic state A2Pi.
For the spin-orbit-x case (-representation), the value of the matrix elements of the 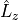 operator must be specified using the
operator must be specified using the <x|Lz|y> keyword. This representation is designed to work with e.g., the MOLPRO outputs. For 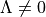 , the diagonal SO-matrix element (e.g. between to
, the diagonal SO-matrix element (e.g. between to 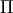 -components of
-components of 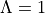 ) should be specified using the
) should be specified using the 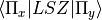 component (e.g.
component (e.g. 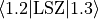 ).
).
spin-spin¶
Parametrised phenomenological spin-spin operator (diagonal and off-diagonal). The diagonal spin-spin matrix elements are given by
.
Note
The definition of 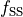 is different from the spectroscopic spin-spin constant
is different from the spectroscopic spin-spin constant 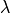 :
:
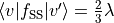 .
.
The non-diagonal spin-spin matrix elements are given by

where 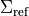 is a reference curve of the projection of spin used to specify the spin-spin field in the Duo input:
is a reference curve of the projection of spin used to specify the spin-spin field in the Duo input:

and  is an off-diagonal spin-spin curve, which is usually reconstructed empirically.
is an off-diagonal spin-spin curve, which is usually reconstructed empirically.
An example of the spin-spin input is given by
spin-spin A a
name "<A|SS|a>"
spin 2.5 1.5
factor 1.0
lambda 0 0
sigma 0.5 0.5
type BOBLEROY
values
RE 0.16500000000000E+01
RREF -0.10000000000000E+01
P 0.10000000000000E+01
NT 0.20000000000000E+01
B0 0.74662463783234E-01
B1 0.73073583911575E+01
B2 0.00000000000000E+00
BINF 0.00000000000000E+00
end
spin-rot¶
The diagonal matrix elements of the spin-rotational operator are given by
.
The nonzero off-diagonal matrix elements are
.
and
.
bob-rot¶
Alias: bobrot and Alpha. Specifies the (diagonal) rotational  factor (rotational Born-Oppenheimer breakdown term),
which can be interpreted as a position-dependent modification to the rotational mass and is introduced as follows
factor (rotational Born-Oppenheimer breakdown term),
which can be interpreted as a position-dependent modification to the rotational mass and is introduced as follows
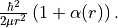
bob-vib¶
Alias: bobvib and Beta. Specifies the (diagonal) vibrational factor (Born-Oppenheimer breakdown term),
which can be interpreted as a position-dependent modification to the vibrational mass and is introduced as follows
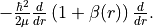
diabatic¶
Alias: diabat. Non-diagonal coupling of potential energy functions in the diabatic
representation. A diabatic coupling should be centred about the crossing point of the correpsonding diabatic potential curves.
For an analitycal (non-grid) representaion, Duo will automatically finds a crossing between the corresponding
states and store its value to the second parameter of the diabatic field. It is threfore important to reserve the second
line for the reference, expansion point. The search of the crossing point is done by the dividing-by-half approach until the
convergence (or 100 iterations) is reached. Only one crossing is currenly supported.
Example:
diabatic B D
name "<B2Sigma+|DC|D2Sigma+>"
lambda 0 0
spin 0.5 0.5
type Lorentz
factor 1.0
values
V0 0.000000000000000000
RE 2.08 (this value will be replaced by the actual crossing point between B and D)
gamma 1.99627265568284e-01
a 2.75756224068962e+02
f1 0.000000000000000000
end
Non-adiabatic coupling: NAC¶
Non-adiabatic coupling (NAC). It is a non-diagonal coupling element used for adiabatic representation. It appears in the kinetic energy operator as a linear momentum term:
,
where 12 stands for the coupling between states 1 and 2. By default, a NAC field trigers the “second order NAC” corrections to the corresponding potential energies defined as
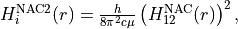
where 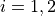 . In Duo, the diagonal
. In Duo, the diagonal diabatic fields are used to store 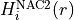 . If however, the corresponding diabatic fields are
directly specified, these second order NAC correction are ignored.
. If however, the corresponding diabatic fields are
directly specified, these second order NAC correction are ignored.
A typical NAC is a Lorentz- or Gaussian-type functions. NAC should be centred about the crossing point of the correpsonding diabatic potential curves.
Example:
NAC B D
name "<B2Sigma+|NAC|D2Sigma+>"
lambda 0 0
spin 0.5 0.5
type Lorentz
factor 1.0
values
V0 0.000000000000000000
RE 2.08 (this value will be replaced by the actual crossing point between B and D)
gamma 1.99627265568284e-01
a 1.0
f1 0.000000000000000000
end
The second order NAC corrections can be provided as two diagonal diabatic fields, e.g. (from the YO spectroscopic model)
Example:
diabatic B B
name "<B2Sigma+|NAC2|B2Sigma+>"
lambda 0 0
spin 0.5 0.5
type grid
factor 1.243548973
values
1.81020 0.0731621425
1.81040 0.0735930439
1.81060 0.0740271189
1.81080 0.0744643954
1.81100 0.0749049019
1.81120 0.0753486669
1.81140 0.0757957194
1.81160 0.0762460887
1.81180 0.0766998042
1.81200 0.0771568959
1.81220 0.0776173938
1.81240 0.0780813285
1.81260 0.0785487308
1.81280 0.0790196317
end
diabatic D D
name "<D2Sigma+|NAC2|D2Sigma+>"
lambda 0 0
spin 0.5 0.5
type grid
factor 1.243548973
values
1.81020 0.0731621425
1.81040 0.0735930439
1.81060 0.0740271189
1.81080 0.0744643954
1.81100 0.0749049019
1.81120 0.0753486669
1.81140 0.0757957194
1.81160 0.0762460887
1.81180 0.0766998042
1.81200 0.0771568959
1.81220 0.0776173938
1.81240 0.0780813285
1.81260 0.0785487308
1.81280 0.0790196317
end
Here factor 1.243548973 is 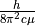 for YO.
for YO.
lambda-opq, lambda-p2q, and lambda-q¶
- These objects are three Lambda-doubling objects which correspond to
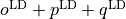 ,
, 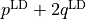 , and
, and 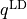 couplings.
couplings.
Example:
lambda-p2q 1 1
name "<X,2Pi|lambda-p2q|X,2Pi>"
lambda 1 1
spin 0.5 0.5
type BOBLEROY
factor 1.0
values
RE 0.16200000000000E+01
RREF -0.10000000000000E+01
P 0.10000000000000E+01
NT 0.20000000000000E+01
B0 0.98500969657331E-01
B1 0.00000000000000E+00
B2 0.00000000000000E+00
BINF 0.00000000000000E+00
end
L2¶
Alias: L**2. These objects represent matrix elements between electronic states of the molecule-fixed angular momentum operator 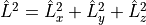 .
.
dipole and dipole-x¶
Dipole (aliases: dipole-moment, TM): Diagonal or transition dipole moment curves (DMCs), necessary for computing (dipole-allowed) transition line intensities and related quantities (Einstein 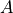 coefficients etc.).
coefficients etc.).
dipole-x is related to the Cartesian-representation (see below).
quadrupole¶
The keyword quadrupole is used to specify transition quadrupole moment curves, which are necessary for computing electric-quadrupole transition line intensities and related quantities. The actual calculation of line strengths requires the quadrupole keyword in the intensity section also (see here).
The quadrupole moment is defined in Cartesian coordinates by the following expression the Shortley convention:
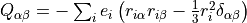
where 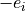 is the charge of the
is the charge of the 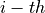 electron with position vector
electron with position vector 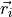 . This differs from the Buckingham convention, which is used in many quantum chemistry programs, where:
. This differs from the Buckingham convention, which is used in many quantum chemistry programs, where:
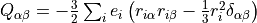
Currently Duo requires quadrupole moment curves to be provided in the spherical irreducible representation, with atomic units (a.u.), which can be obtain from the Cartesian components in the Buckingham convention via the relations given by Eq. (6) - (11) of W. Somogyi et al., JCP 155, (2021).
Additionally, the units must be specified via the units keyword. For example
quadrupole 1 1
name "<X3Sigma-|QM20|X3Sigma->"
spin 1 1
lambda 0 0
type grid
units angstrom au
values
0.8 -1.4747
0.9 -1.1434
...
end
MAGNETIC¶
Aliases: MAGNETIC-DIPOLE,``MAGNETIC-X``. Specifies the magnetic dipoles used in magnetic dipole intensity calculations, its electronic orbital angular momentum contribution 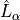 :
.. math:
:
.. math:
\mu_\alpha = \hat{S}_\alpha + \hat{L}_\alpha,
where 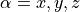 . If
. If Magnetic fields are not provided for magdipole intensity calculations, the corresponding fields Lx are used instead.
MAGNETROT¶
Aliases: MAGNETIC-ROT,``MAGNET-ROT``. Specifies the rotational magnetic dipoles used in magnetic dipole intensity calculations. This contribution corresponds to the rotational g-factor, which is the same as the BOB-ROT component. If MagnetRot fields are not provided for magdipole intensity calculations, the corresponding fields Bob-Rot are used instead.
Warning
This feature is currently under construction.
Keywords used in the specification of objects¶
Name and quantum numbers¶
This is a list of keywords used to specify various parameters of Duo objects.
name: object name.
name is a text label which can be assigned to any object for reference in the output. The string must appear within quotation marks.
Examples:
name "X 1Sigma+"
name "<X1Sigma\|HSO\|A3Pi>"
lambda: The quantum number(s).
Lambda specifies the quantum number(s) , i.e. projections of the electronic angular momentum onto the molecular axis, either for one (PECs) or two states (couplings). It must be an integral number and is allowed to be either positive or negative. The sign of is relevant when specifying couplings between degenerate states in the spherical representation (e.g. spin-orbit)
Examples:
lambda 1
lambda 0 -1
The last example is relative to a coupling-type object and the two numbers refer to the bra and ket states.
sigma: Spin-projection.
sigma specifies the quantum number(s) 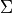 , i.e. the projections of the total spin onto the molecular axis, either for one (diagonal) or two states (couplings). These values should be real (
, i.e. the projections of the total spin onto the molecular axis, either for one (diagonal) or two states (couplings). These values should be real (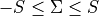 ) and can be half-integral, where
) and can be half-integral, where 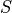 is the total spin.
is the total spin. sigma is currently required for the spin-orbit couplings only.
Example:
sigma 0.5 1.5
where two numbers refer to the bra and ket states.
mult(alias:multiplicity): Multiplicity
mult specifies the multiplicity of the electronic state(s), given by 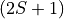 , where is the total spin. It must be an integer number and is an alternative to the
, where is the total spin. It must be an integer number and is an alternative to the spin keyword.
Examples:
mult 3
mult 1 3
The last example is relative to a coupling-type object and the two numbers refer to the bra and ket states.
spin: Total spin.
The total spin of the electronic state(s), an integer or half-integer number.
Example:
spin 1.0
spin 0.5 1.5
The last example is relative to a coupling-type object and the two numbers refer to the bra and ket states.
symmetry: State symmetry
This keyword tells Duo if the electronic state has gerade g or ungerade u symmetry (only for homonuclear diatomics) and whether it has positive (+) or negative - parity (only for states, i.e. states with 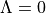 , for which it is mandatory).
, for which it is mandatory).
Examples:
symmetry +
symmetry + u
symmetry g
The keywords g/u or +/- can appear in any order.
Other control keys¶
type: Type of the functional representaion.
Type defines if the object is given on a grid type grid or selects the parametrised analytical function used for representing the objects or selects the interpolation type to be used. The function types supported by Duo are listed in Duo Functions.
Examples:
type grid
type polynomial
type morse
In the examples above grid selects numerical interpolation of values given on a grid, polynomial selects a polynomial expansion and morse selects a polynomial expansion in the Morse variable. See Duo Functions for details.
Interpolationtype: Grid interpolation
is used only for type grid and specifies
the method used for the numerical interpolation of the numerical values. The currently implemented interpolation methods are Cubicsplines and Quinticsplines (default).
Example:
Interpolationtype Cubicsplines
Interpolationtype Quinticsplines
factor: Scaling factor
This optional keyword permits to rescale any object by an arbitrary multiplication factor. At the moment the accepted values are any real number, the imaginary unit 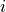 , the square root of two, written as
, the square root of two, written as sqrt(2), or products of these quantities. To write a product simply leave a space between the factors, but do not use the * sign. All factor can have a 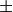 sign. The default value for
sign. The default value for factor is 1. This keyword is useful, for example, to temporarily zero a certain object without removing it from the input file.
Examples:
factor 1.5
factor -sqrt(2)
factor sqrt(2)
factor 5 i
factor -2 sqrt(2) i
In the last example the factor is read in as 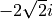 . Note that imaginary factors make sense only in some cases for some coupling terms (in particular, spin-orbit) in the Cartesian-representation, see Section~ref{s:representations}.
. Note that imaginary factors make sense only in some cases for some coupling terms (in particular, spin-orbit) in the Cartesian-representation, see Section~ref{s:representations}.
units
This keyword selects the units of measure used for the the object in question. Supported units are: angstroms (default) and bohr for the bond lengths; cm-1 (default), hartree (aliases are au, a.u., and Eh), and eV (electronvolts) for energies; debye (default) and ea0 (i.e., atomic units) for dipoles; units can appear in any order.
Note
Quadrupole moment curves must be provided to Duo in atomic units, so the units keyword is invalid for these objects.
Example:
units angstrom cm-1 (default for poten, spin-orbit, lambda-doubling etc)
units bohr cm-1
units debye (default)
units ae0 bohr
<x|Lz|y>,<z|Lz|xy>(aliases<a|Lz|b>and<1|Lz|2>)
This keyword is sometimes needed when specifying coupling curves between electronic states with  in order to resolve ambiguities in the definition of the degenerate components of each electronic state, see:ref:representations.
in order to resolve ambiguities in the definition of the degenerate components of each electronic state, see:ref:representations.
This keyword specifies the matrix element of the operator between the degenerate components of the electronic wave function.
Examples:
<x|Lz|y> i -i
<z|Lz|xy> -2i i
These matrix elements are pure imaginary number in the form 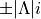 . It is the overall sign which Duo needs and cannot be otherwise guessed. As shown in the examples above, each factor should be written in the form without any space or
. It is the overall sign which Duo needs and cannot be otherwise guessed. As shown in the examples above, each factor should be written in the form without any space or * sign.
Molpro
A single, stand-alone keywrd to trigger the molpro even for non-x fields.
Example:
molpro
morphing
This keyword is used for fitting and switches on the morphing method.
ZPE: Zero-point-energy
ZPE allows to explicitly input the zero-point energy (ZPE) of the molecule (in cm-1). This affects the value printed, as by default Duo prints energy of rovibronic levels by subtracting the ZPE. If not specified, the lowest energy of the first 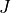 -block (independent of parity) will be used as appear on the line
-block (independent of parity) will be used as appear on the line Jlist.
fit_factor
This factor (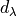 ) is used as a part of the reference ab initio curves of the
) is used as a part of the reference ab initio curves of the abinitio type which (when given) is applied to the corresponding weights assigned to the corresponding values of this object. It is different from fit_factor defined within in Duo fitting.
adjust
This keyword can be used to add a constant value to the values of the potential, which is useful e.g when there is a known systematic error in the values. The keyword is followed by a value and (optionally) units. For a list of the available units see the units keyword above. Note that the units of the shift can be different to the units specified using the units keyword. Default units are cm-1 for PECs, debye for dipole moment curves, and au (atomic units) for quadrupole moment curves.
Examples:
adjust -42 cm-1
adjust
Example:
abinitio poten 1
name "A 1Pi"
type grid
lambda 1
mult 1
units bohr cm-1
fit_factor 1e1
values
2.00 32841.37010 0.01
2.20 17837.88960 0.10
2.40 8785.33147 0.70
2.60 3648.35520 1.00
2.70 2107.10737 1.00
2.80 1073.95670 1.00
2.90 442.52180 1.00
3.00 114.94960 1.00
3.10 0.00000 1.00
3.20 48.46120 1.00
3.30 213.34240 1.00
3.40 455.16980 1.00
3.50 739.61170 1.00
3.60 1038.82620 1.00
3.70 1332.46170 1.00
4.00 2059.31119 1.00
4.50 2619.19233 0.30
5.00 2682.84741 0.30
6.00 2554.34992 0.30
8.00 2524.31106 0.30
10.00 2561.48269 1.00
12.00 2575.09861 1.00
end
Cartesian or tensorial representation¶
Internally, Duo represents electronic basis functions in Hund’s case (a), where the electronic function
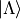 is an eigenfunction of ,
is an eigenfunction of ,
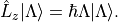
In this Lambda-representation (also referred to as the tensorial representation), the  component is diagonal
and carries the signed value . Duo therefore stores and manipulates coupling matrix elements and transition
moments in this tensorial form. For example, the electronic angular momentum ladder operators are
component is diagonal
and carries the signed value . Duo therefore stores and manipulates coupling matrix elements and transition
moments in this tensorial form. For example, the electronic angular momentum ladder operators are
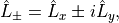
and the spherical (tensorial) components of the body-fixed dipole moment are
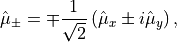
where , 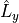 ,
, 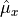 , and
, and 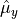 are Cartesian components.
are Cartesian components.
Most quantum-chemistry programs (for example Molpro [1]) use real electronic wavefunctions and compute
matrix elements of multi-component properties (spin–orbit, electronic angular momentum, dipole moments, etc.) in a
Cartesian representation. In this representation, electronic functions are ultimately expressed in terms of real
Cartesian components (e.g. [2]) 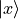 ,
, 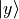 ,
, 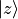 ,
,
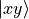 , etc.
, etc.
It can be shown that, with appropriate phase choices for the complex Hund’s-case-(a) functions
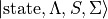 , all coupling matrix elements in the Lambda-representation can be made real.
Note that the underlying electronic functions in the Lambda-representation are complex (they contain factors of the form
, all coupling matrix elements in the Lambda-representation can be made real.
Note that the underlying electronic functions in the Lambda-representation are complex (they contain factors of the form
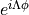 , where
, where 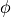 is the azimuthal angle about the molecular axis).
is the azimuthal angle about the molecular axis).
For that reason, the Lambda-representation is the native representation for Duo. The corresponding input objects assume
tensorial components, for example dipole, spin-orbit, L+/L- (and related operators), etc.
Cartesian input¶
Duo can also accept input in the Cartesian representation, which is often the most direct output of quantum-chemistry calculations. In this case Duo assumes that both the electronic wavefunctions and the property operator are given in the Cartesian representation. For many multi-component properties, only one Cartesian component is required (a reference component), since all other non-zero matrix elements can be generated by symmetry/selection rules. Therefore, for Cartesian input Duo typically requires only the x component.
In this case, use the *-x object names, e.g. dipole-x, spin-orbit-x and Lx.
Transformation to the Lambda-representation¶
For Cartesian input, Duo transforms the supplied matrix elements to the Lambda-representation on the fly using a
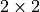 unitary transformation,
unitary transformation,
(1)¶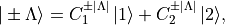
where 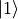 and
and 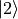 denote the relevant Cartesian components (e.g.
denote the relevant Cartesian components (e.g. 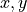 or other real
combinations such as
or other real
combinations such as 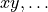 ) corresponding to Abelian point-group symmetry pairs (e.g.
) corresponding to Abelian point-group symmetry pairs (e.g. 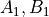 or
or
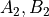 ). The coefficients
). The coefficients 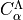 are elements of the unitary transformation from the
Cartesian basis to the Lambda-representation.
are elements of the unitary transformation from the
Cartesian basis to the Lambda-representation.
For non-diagonal spin–orbit couplings with 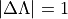 reported by Molpro, the relevant Cartesian reference matrix element is an
reported by Molpro, the relevant Cartesian reference matrix element is an 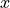 component, e.g.
component, e.g. 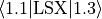 (use
(use spin-orbit-x). In the Lambda-representation the corresponding matrix element acquires a factor of 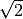 .
.
Note
Do not mix representations within the same object. If the electronic wavefunctions and property are taken from a
Cartesian quantum-chemistry calculation, use the corresponding *-x objects consistently (e.g. spin-orbit-x,
dipole-x).
Note
For perpendicular dipole transitions with , the Cartesian component (dipole-x),
e.g. 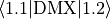 , has the same numerical value as the tensorial body-fixed
component used internally by Duo. The factor from the basis transformation is cancelled by the
, has the same numerical value as the tensorial body-fixed
component used internally by Duo. The factor from the basis transformation is cancelled by the
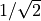 in the definition of
in the definition of 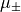 above. Therefore, do not apply any manual
conversion for dipoles;
above. Therefore, do not apply any manual
conversion for dipoles; dipole and dipole-x accept the same numerical values and Duo handles
the transformation internally.
Definition of the function or a grid¶
values
This keyword starts the subsection containing the numerical values defining the object. For one of the type``s corresponding to an analytic function, the input between ``values and end contains the values of the parameters of the function. The input consists in two columns separated by spaces containing (i) a string label identifying the parameter and (ii) the value of the parameter (a real number).
In case of fitting (see Duo fitting) a third column should also be provided; the parameters which are permitted to vary during fitting
must have in the third column the string fit or, alternatively, the letter f or the number 1. Any other string or number (for example, the string nofit or the number 0) implies the parameter should be kept at its initial value. In the case of fitting, the keyword link
can be also appear at the end of each the line; this keyword permits to cross-reference values from different objects and is explained
below in this section.
In the case of objects of type grid, the third column can be also used to specify if the grid point needs to vary. The first columns contains the bond length 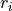 and a second with the value of the object. In the case of object of the
and a second with the value of the object. In the case of object of the abinitio (reference) type and specified as grid a third column can be used to specify the fitting weights (see Duo fitting).
link
This special keyword is used in fitting to force a set of parameters (which may be relative to a different object) to have the same value. For example, in a typical situation one may want to fit a set of PECs and to constrain their dissociation (asymptotic) energy to the same value (because they are expected from theory to share the same dissociation channel).
After the keyword link one should provide three numbers 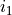 ,
, 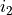 ,
, 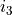 defining the parameter ID, where identifies the object type (e.g.
defining the parameter ID, where identifies the object type (e.g. poten, spin-orbit, spin-rot etc.), is the object number within the type and is the parameter number as it appears after values. The ID numbers 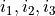 are specified in the fitting outputs in the form [i,j,k].
are specified in the fitting outputs in the form [i,j,k].
Example of the input:
DE 0.50960000000000E+05 fit link 1 1 3
Example of the corresponding output
DE 0.50960000000000E+05 [ 1 1 3 ]
Warning
The link feature, where the associated value is replaced with the value the associated parameter is linked to, currently only works when the fitting is on. For all other cases the link keyword is ignored and the actual input value of the given parameter is used, not the one it is linked to. Therefore, to be on the safe side, the correct (fitted) value must be transferred to the place next to the parameter.
Using ab initio couplings in Duo: Representations of the electronic wave functions¶
Quantum chemistry programs generally use real-valued electronic wave functions which transform according to the irreducible representations of the C:sub:2v point group (for heteronuclear diatomics) or of D:math:2h (for homonuclear diatomics). On the other hand Duo internally assumes the electronic wave functions are eigenfunctions of the operator, which implies they must be complex valued for . Converting from one representation to the other is simple, as
![|\Lambda\rangle =\frac{1}{\sqrt{2}}\left[\mp |1\rangle - i|2\rangle \right].](_images/math/3da34e40c39fa2ecc0117eab7d06175dfb3e53a3.png)
where  and
and  are two Cartesian components of the electronic wave functions in a quantum chemistry program. Duo uses the matrix elements of the to reconstruct the transformation between two representations:
are two Cartesian components of the electronic wave functions in a quantum chemistry program. Duo uses the matrix elements of the to reconstruct the transformation between two representations:
The keyword <x|Lz|y> and <z|Lz|xy> (aliases <a|Lz|b> and <1|Lz|2>) is required when specifying coupling curves between electronic states in the MOLPRO representation (spin-orbit-x, Lx and dipole-x) with in order to resolve ambiguities in the definition of the degenerate components of each electronic state. This is also the value of the matrix element of the operator computed for the two component spherical harmonic, degenerate functions and for the states or and for the 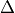 states etc. The corresponding <x|Lz|y> values for both coupled states must be provided.
states etc. The corresponding <x|Lz|y> values for both coupled states must be provided.
Examples:
<x|Lz|y> i -i
<z|Lz|xy> -2i i
This keyword is required for the couplings of the following types: spin-orbit-x, Lx and dipole-x. The suffix -x indicates that Duo expects the x-component (non-zero) of the corresponding coupling.
This keyword should appear anywhere in the object section, before the values keyword.
spin-orbit-x 1 1
name "X-X SO term"
spin 1.0 1.0
lambda 2 2
sigma 1.0 1.0
units angstrom cm-1
type polynomial
factor i
<x|Lz|y> 2i 2i
values
f 101.2157
end
These matrix elements are pure imaginary number in the form . It is the overall sign which Duoneeds and cannot be otherwise guessed. As shown in the examples above, each factor should be written in the form without any space or * sign.